MD Simulations
Although experimental methods are inevitable for unraveling the behavior of bio macro molecules, they mostly lack atomic detail. Here, molecular dynamics (MD) simulations are the method of choice to complement the experimental results. Due to the size of the molecules under study our "working horses" are programs that employ Newtonian mechanics with classical potentials for the nuclei but in special cases we are using also quantum mechanical methods as well as combined quantum mechanical and molecular mechanical (QM/MM) programs and Monte Carlo (MC) simulations.
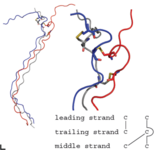
An example for the application of purely classical molecular dynamics is the investigation of a cystein rich region of collagen. For this large protein a series of different models have been simulated in order to gain more information about its possible structure (see Fig. 1 on the right, taken from Ref. 1).
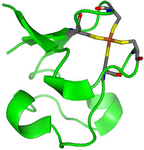
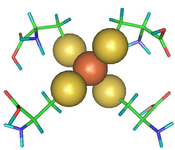
A case where a QM/MM approach has been chosen is given by rubredoxin, an enzyme with a catalytically active iron-sulfur center, that had to be described within a quantum mechanical framework, while the remaining part of the protein was treated with a more affordable technique (see Fig. 2 and 3 taken from Ref. 2).
Since cooperativity is central to folding and function of proteins, we are in general very interested in this topic. Small (in comparison to bio molecules) model systems, that allow to study cooperativity, are spin crossover compounds. In a recent survey we have used a quantum mechanical approach to contribute to the unstanding of cooperativity in these compounds.

Hauke Paulsen

Gebäude 61
,
Raum 221
paulsen(at)physik.uni-luebeck.de
+49 451 3101 4203